甲烷,作為天然氣的主要成分,不僅是一種清潔的能源,也是合成多種高附加值化學品的重要原料。然而,由于其分子結構的穩(wěn)定性,甲烷的化學轉化過程通常需要高溫高壓條件,這不僅消耗大量能源,還可能引發(fā)環(huán)境問題。為了解決這一挑戰(zhàn),光催化技術應運而生,它利用太陽能在溫和條件下激活甲烷,為化學合成提供了一種可持續(xù)的解決方案。盡管這一領域充滿潛力,但目前仍面臨諸多技術難題。
在傳統的甲烷轉化過程中,如蒸汽甲烷重整,雖然實現了甲烷的大規(guī)模工業(yè)應用,但這一過程不僅能耗高,還伴隨著大量的二氧化碳排放。因此,探索在溫和條件下直接轉化甲烷的方法,對于提高能源利用效率和減少環(huán)境污染具有重要意義。[1-3]
近年來,光催化技術因其在溫和條件下活化甲烷的能力而受到廣泛關注。光催化甲烷轉化利用光能激發(fā)催化劑,產生高能電子-空穴對,這些高能電子-空穴對能夠打破甲烷分子的化學惰性,從而在較低的溫度和壓力下實現甲烷的高效轉化。[4-6]這一過程不僅能夠降低能耗,還能減少溫室氣體排放,對于推動綠色化學合成具有重要的戰(zhàn)略意義。
甲烷以其高度的化學穩(wěn)定性而著稱,這種穩(wěn)定性源自其對稱的四面體幾何結構,其中四個等價的C-H鍵賦予了它高鍵能、低電子及質子親和力和低極化性(見圖1a)。[7,8]此外,甲烷的最高占據分子軌道(HOMO)與最低未占據分子軌道(LUMO)之間的能隙較大,這增加了它在分子層面上獲得或失去電子的難度。[1,9]這種內在的化學惰性意味著在熱催化轉化過程中,需要逾越較高的活化能障礙(Ea),通常需要在700至1000攝氏度的高溫下進行,以實現有效的轉化。盡管多年來科學家們不懈努力開發(fā)新型催化劑以提高甲烷轉化的效率,但熱催化過程的高能耗和碳排放問題依然存在。目前,唯一大規(guī)模應用的甲烷轉化技術是將其通過蒸汽重整間接轉化為合成氣,進而生產烯烴、甲醇和液態(tài)烴。而將甲烷高效轉化為一系列含碳化學品,包括醇類、芳香烴、長鏈烷烴和烯烴,一直是催化化學領域追求的目標。[10]
非均相光催化劑繼電子-空穴分離和轉移過程之后,主要涉及催化劑-介質界面上發(fā)生的過程,包括反應物的吸附與活化、中間體的生成以及產物的解吸與脫附等。[11-13]催化界面上的復雜反應過程對精細調節(jié)催化反應的總效率提出了挑戰(zhàn)。[14,15]甲烷活化通常被認為是甲烷轉化反應中的速率控制步驟。[16,17]基于非均相催化劑的光催化甲烷活化可分為兩類:
1)直接活化,即甲烷在光照條件下直接吸附在光催化劑表面被活化(圖 1d);
2)間接活化,即甲烷在水和氧分子等其他反應物的光誘導活性自由基的幫助下被活化(圖 1e)。
盡管兩種甲烷活化途徑之間存在差異,但光誘導產生的活性氧,如產生光生表面活性位點(氧化物上的O?)和活性自由基(•OH和•O²?)在以上敘述的兩種途徑中都發(fā)揮著關鍵作用。
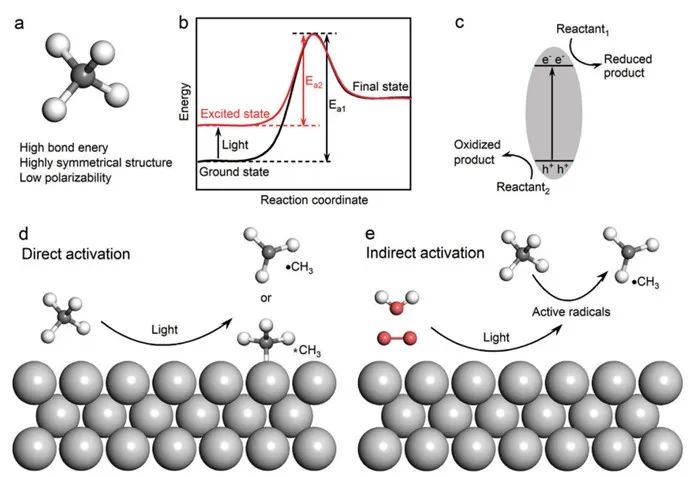
圖1 a)甲烷的分子結構。b)基態(tài)和激發(fā)態(tài)反應中的能量變化。c)光催化反應的基本原理。d)甲烷在光催化劑表面的直接活化。e)光誘導活性自由基對甲烷的間接活化。
對于直接活化途徑而言,為優(yōu)化光催化甲烷活化,通常改性策略是酸/堿位點和晶體缺陷工程改性策略。[18-20]金屬氧化物上的堿性位點被認為在甲烷活化中起著至關重要的作用,因為它們與弱酸性甲烷具有相對較強的電子相互作用。[21-23]吸附在金屬氧化物上的甲烷會形成與金屬陽離子配位的負電基團和與金屬氧化物的堿性晶格氧配位的正電基團,二者作用使金屬氧化物表面的 C–H 鍵極化(圖 2a)。根據金屬氧化物的堿性強弱,這兩部分將顯示路易斯酸(正)或路易斯堿(負)。[24]此外,納米催化劑中的缺陷導致晶胞平移對稱性的破壞,包括三維體積缺陷(如孔隙)、二維平面缺陷(如晶界)、一維線缺陷(位錯)和零維點缺陷(如空位)。這些缺陷直接影響金屬氧化物的配位結構和相應的催化性能。[25]例如,Pd修飾的ZnO-Au復合材料具有獨特的界面缺陷結構,促進CH?分子解離為甲氧基和甲基,促進光催化甲烷-乙烯轉化(圖2b)。[26]
對于間接途徑來說,氫原子轉移是激活甲烷 C–H 鍵的有效策略。它是涉及通過自由基反應中質子和電子的分離而轉移氫原子(圖 2c)的一個基本反應。[27,28]光催化氫原子轉移是利用光催化過程中產生的自由基中間體來活化 C–H 鍵。[29]氣相中的裸露的 (CuO)? 陽離子可以與以氧為中心的自由基相互作用,促進氫原子轉移和甲烷活化。[30]據報道,基于同步輻射的原位光催化質譜技術可捕獲光催化甲烷氧化反應中的氣相活性中間體。[31]除了能夠檢測到 CO?、H?O、C?H? 和 CH?OH 等幾種穩(wěn)定物質外,還能夠檢測到活性甲基自由基(•CH?)和甲氧基自由基(CH?O•)(圖 2d)。此外,質譜結合量子化學計算顯示,(AuO)? 通過選擇性地從 (AuO)? 中轉移氧原子,而不是從甲烷中提取氫原子轉移氫原子,將甲烷活化為甲醇,盡管在同系物 (CuO)? 和 (AgO)? 中觀察到了后一種情況。
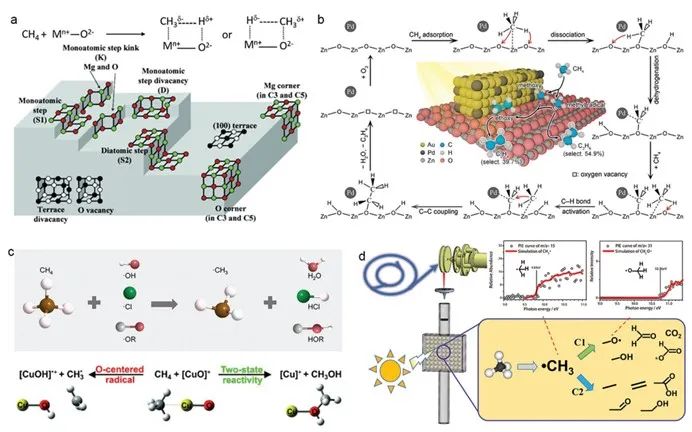
圖 2. a) 甲烷在堿性金屬氧化物表面的吸附和 C–H 鍵極化以及 MgO 表面不同位置的示意圖。[21,22] b) 在 ZnO-AuPd 混合催化劑的情況下,通過表面烷氧基中間體將 CH? 光催化轉化為 C?H? 的示意圖。[26] c) 通過氫原子轉移活化甲烷的示意圖。[30] d) 通過原位同步輻射光電離質譜法檢測光催化甲烷氧化反應過程中的反應中間體。[31]
甲烷的氧化脫氫反應是另一種廣泛研究的溫和條件下甲烷活化和轉化的間接途徑[32]。根據涉及到的氧活性物質的形式,甲烷的氧化脫氫反應主要遵循三種機理:Rideal-Eley (R-E)機理和Langmuir-Hinshelwood (L-H)機理,二者均以表面吸附氧為主,以及Mars-van Krevelen (Mv-K)機理,是以晶格氧為主。下面簡單介紹一下這些反應機理:
1)R-E機理:CH?分子最初與過渡金屬氧化物中的晶格氧結合,導致CH?分子中的C–H鍵斷裂并形成自由甲基自由基,隨后甲基自由基被氧化。[33]同時,結合過程會消耗晶格中的氧,從而形成弱電氧空位。這些空位吸收分子氧來補充失去的晶格氧,從而完成催化循環(huán)(圖3a)。
2)L-H機理:催化劑表面對分子氧的化學吸附明顯比對甲烷的化學吸附容易,這導致分子氧在暴露于含氧大氣中時優(yōu)先吸附在催化劑表面[34]與分子氧相比,這些被吸附的氧物種與CH?分子結合時表現出更高的反應活性,導致甲烷中C–H鍵被拉長。這種拉長隨后會破壞了CH?分子的正四面體結構,產生活性甲基自由基,促進 CH?的氧化脫氫(圖3b)。L–H 機理主要用于貴金屬負載的金屬氧化物催化劑。[35]
3)Mv-K機理涉及表面氧反應和晶格氧遷移。[36]首先,氣態(tài)CH?分子被吸附在催化劑的活性位點上。然后,吸附的CH?分子與表面晶格氧發(fā)生反應,生成 CH?? 離子,隨后被氧化成 CO? 和 H?O 分子等吸附產物,從而產生氧空位。這一步驟可稱為催化劑還原。最后,吸附產物被解吸,而內部晶格氧遷移到表面,用表面吸附的氧重新填滿氧空位,這就是催化劑的再氧化(圖 3c)。
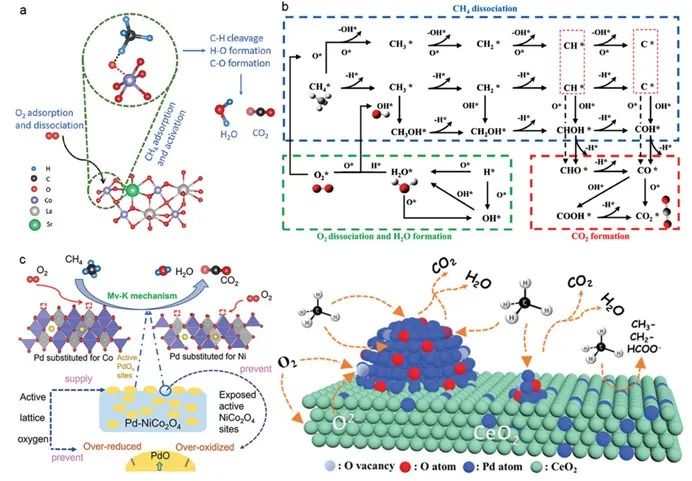
圖3. a) 針對鑭、鈷基鈣鈦礦氧化物催化劑提出的超面 Rideal-Eley 機理示意圖。[33]b) 通過 Langmuir-Hinshelwood 機理進行的甲烷催化燃燒反應途徑。[35] c) 通過 Mars-van Krevelen 機理進行的甲烷氧化反應,其中 Pd 摻入 Pd-NiCo?O? 和 Pd/CeO?。[36,37]


參考文獻
[1]. N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi, R. A. Periana, Chem. Rev. 2017, 117, 8521.
[2]. Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi, C. T. Yavuz, Science 2020, 367, 777.
[3]. Y. Chen, J. Wei, M. S. Duyar, V. V. Ordomsky, A. Y. Khodakov, J. Liu, Chem. Soc. Rev. 2021, 50, 2337.
[4]. T. Kong, Y. Jiang, Y. Xiong, Chem. Soc. Rev. 2020, 49, 6579.
[5]. P. Bellotti, H.-M. Huang, T. Faber, F. Glorius, Chem. Rev. 2023, 123, 4237.
[6]. L. Zhang, L. Liu, Z. Pan, R. Zhang, Z. Gao, G. Wang, K. Huang, X. Mu, F. Bai, Y. Wang, W. Zhang, Z. Cui, L. Li, Nat. Energy 2022, 7, 1042
[7]. P. Tang, Q. Zhu, Z. Wu, D. Ma, Energy Environ. Sci. 2014, 7, 2580.
[8]. B. Wang, S. Albarracín-Suazo, Y. Pagán-Torres, E. Nikolla, Catal. Today 2017, 285, 147.
[9]. H. Schwarz, Angew. Chem., Int. Ed. 2011, 50, 10096.
[10]. R. G. Bergman, Nature 2007, 446, 391.
[11]. J. C. Védrine, Top. Catal. 2002, 21, 97.
[12]. I. Fechete, Y. Wang, J. C. Védrine, Catal. Today 2012, 189, 2.
[13]. R. Schlögl, Angew. Chem., Int. Ed. 2015, 54, 3465.
[14]. C. M. Friend, B. Xu, Acc. Chem. Res. 2017, 50, 517.
[15]. X. Cui, W. Li, P. Ryabchuk, K. Junge, M. Beller, Nat. Catal. 2018, 1, 385.
[16]. Y. Chen, X. Mu, X. Luo, K. Shi, G. Yang, T. Wu, Energy Technol. 2020, 8, 1900750.
[17]. S. H. Morejudo, R. Zanón, S. Escolástico, I. Yuste-Tirados, H. Malerød-Fjeld, P. K. Vestre, W. G. Coors, A. Martínez, T. Norby, J. M. Serra, C. Kjølseth, Science 2016, 353, 563.
[18]. M. Campanati, G. Fornasari, A. Vaccari, Catal. Today 2003, 77, 299.
[19]. A. Vojvodic, J. K. Nørskov, Natl. Sci. Rev. 2015, 2, 140.
[20]. A. Nilsson, L. G. M. Pettersson, B. Hammer, T. Bligaard, C. H. Christensen, J. K. Nørskov, Catal. Lett. 2005, 100, 111.
[21]. V. Choudhary, J. Catal. 1991, 130, 411.
[22]. H. U. Hambali, A. A. Jalil, A. A. Abdulrasheed, T. J. Siang, T. A. T. Abdullah, A. Ahmad, D.-V. N. Vo, Int. J. Energy Res. 2020, 44, 5696.
[23]. J. Ashok, Z. Bian, Z. Wang, S. Kawi, Catal. Sci. Technol. 2018, 8, 1730.
[24]. C. Chizallet, G. Costentin, M. Che, F. Delbecq, P. Sautet, J. Phys. Chem. B 2006, 110, 15878.
[25]. W. Jiang, J. Low, K. Mao, D. Duan, S. Chen, W. Liu, C.-W. Pao, J. Ma, S. Sang, C. Shu, X. Zhan, Z. Qi, H. Zhang, Z. Liu, X. Wu, R. Long, L. Song, Y. Xiong, J. Am. Chem. Soc. 2021, 143, 269.
[26]. Y. Jiang, Y. Fan, S. Li, Z. Tang, CCS Chem. 2022, 5, 30.
[27]. B. An, Q.-H. Zhang, B.-S. Zheng, M. Li, Y.-Y. Xi, X. Jin, S. Xue, Z.-T. Li, M.-B. Wu, W.-T. Wu, Angew. Chem., Int. Ed. 2022, 61, e202204661.
[28]. J. Wang, L. Zhang, D. Zeng, W. Wang, R. Li, T. Jia, B. Cui, H. Chu, W. Wang, Appl. Catal., B 2023, 337, 122983.
[29]. N. Dietl, C. Van Der Linde, M. Schlangen, M. K. Beyer, H. Schwarz, Angew. Chem., Int. Ed. 2011, 50, 4966.
[30]. C. Liu, B. Qian, T. Xiao, C. Lv, J. Luo, J. Bao, Y. Pan, Angew. Chem., Int. Ed. 2023, 62, e202304352.
[31]. S. Zhou, J. Li, M. Schlangen, H. Schwarz, Angew. Chem., Int. Ed. 2016, 55, 10877.
[32]. T. Wang, C. Zhang, J. Wang, H. Li, Y. Duan, Z. Liu, J. Y. Lee, X. Hu, S. Xi, Y. Du, S. Sun, X. Liu, J.-M. Lee, C. Wang, Z. J. Xu, J. Catal. 2020, 390, 1.
[33]. X. Bu, J. Ran, J. Niu, Z. Ou, L. Tang, X. Huang, Mol. Catal. 2021, 515, 111891.
[34]. E. Becker, P.-A. Carlsson, L. Kylhammar, M. A. Newton, M. Skoglundh, J. Phys. Chem. C 2011, 115, 944.
[35]. T. Wang, L. Qiu, H. Li, C. Zhang, Y. Sun, S. Xi, J. Ge, Z. J. Xu, C. Wang, J. Catal. 2021, 404, 400.
[36]. S. Chen, S. Li, R. You, Z. Guo, F. Wang, G. Li, W. Yuan, B. Zhu, Y. Gao, Z. Zhang, H. Yang, Y. Wang, ACS Catal. 2021, 11, 5666.
[37]. D. Hu, V. V. Ordomsky, A. Y. Khodakov, Appl. Catal., B 2021, 286, 119913.